Re-reading a 2018 melanoma single-cell atlas with a translational lens: how an exclusion-axis reframing nominates five tier-1 anti-PD-1 combinations
A re-analysis of GSE115978 (Jerby-Arnon et al., Cell 2018) — what a translational team can extract today that the original paper did not.
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175(4):984–997.e24, 1 November 2018 (PMID 30388455). Data: GEO GSE115978 (Smart-seq2 scRNA-seq across 33 melanoma tumours).
- Exclusion-axis reframing. Post-treatment biopsies are NOT enriched among the most excluded tumours (Fisher OR = 0.76, p = 0.77). A per-patient CD8/Mal log-ratio is a sharper resistance stratifier than naive vs post-treatment status — and is what every downstream contrast was anchored to.
- Two orthogonal methods converge on the published Jerby-Arnon programme. Consensus NMF (MP10) and pseudobulk DESeq2 (98-gene consensus) share zero gene-list overlap (hypergeometric p = 1.0) yet both project onto the JA induced/repressed modules at NES = +2.58 / −2.51 — a cross-method internal validation at the pathway level.
- 309 LR pairs enriched in resistance contexts. Mal-as-sender is dominated by an MIF / COPA / APP → CD74 hub. Top Δ-enriched axes: CD72 → SEMA4D (rank 1, macrophage → Mal), RPS19 → C5aR1 complement evasion (4 of top 13), LILRB4 → LAIR1 myeloid checkpoint, and CAF collagen → integrin physical exclusion.
- Five tier-1 anti-PD-1 combinations after safety filtering. Bevacizumab, glembatumumab vedotin, avacopan, pepinemab, plerixafor — three of which (avacopan, pepinemab, plerixafor) map directly to specific LR pairs identified upstream.
- Safety filter flips the shortlist. The two highest composite scorers (CDK4/6i palbociclib/abemaciclib) are demoted on hepatotoxicity grounds; the largest single fold-change in the consensus signature (EDN3) is demoted on bosentan/macitentan boxed warnings.
Why revisit this dataset?
Jerby-Arnon and colleagues established GSE115978 as a foundational scRNA-seq map of the malignant-cell programmes that drive T-cell exclusion in melanoma. Seven years on, with mature ligand–receptor inference, consensus NMF, OpenTargets/DGIdb-grade target intelligence, IMPC knockout phenotypes, and a curated secondary-pharmacology safety panel, the same 32-patient cohort can be pushed considerably further toward a safety-filtered, biology-anchored anti-PD-1 combination shortlist. That is the translational arc of this re-analysis.
We worked from a Harmony-integrated object of 6,879 cells × 22,411 genes balanced across treatment-naive (3,447) and post-treatment (3,432) and across the Tirosh and New cohorts (3,998 / 2,881).
Finding 1 — Reframing "post-treatment" as a heterogeneous group
The single most consequential reframing is methodological. Defining a per-patient exclusion score
and stratifying into tertiles (11 / 10 / 11 patients) shows that post-treatment biopsies are NOT enriched among the exclusion-high tumours (Fisher OR = 0.76, p = 0.77). In fact, 8 of 11 exclusion-low patients are post-treatment — consistent with successful CD8 infiltration in responders — while the most severely excluded tumours (Mel78: 0 CD8 / 120 Mal; Mel106: 8 / 94) are treatment-naive immune deserts.
Translational consequence: "naive vs post" is the wrong axis for resistance biology in this cohort. The exclusion-tertile axis is sharper and was used to anchor every downstream contrast. 1,206 cells were flagged resistance_proxy = True (post-treatment AND exclusion-high).
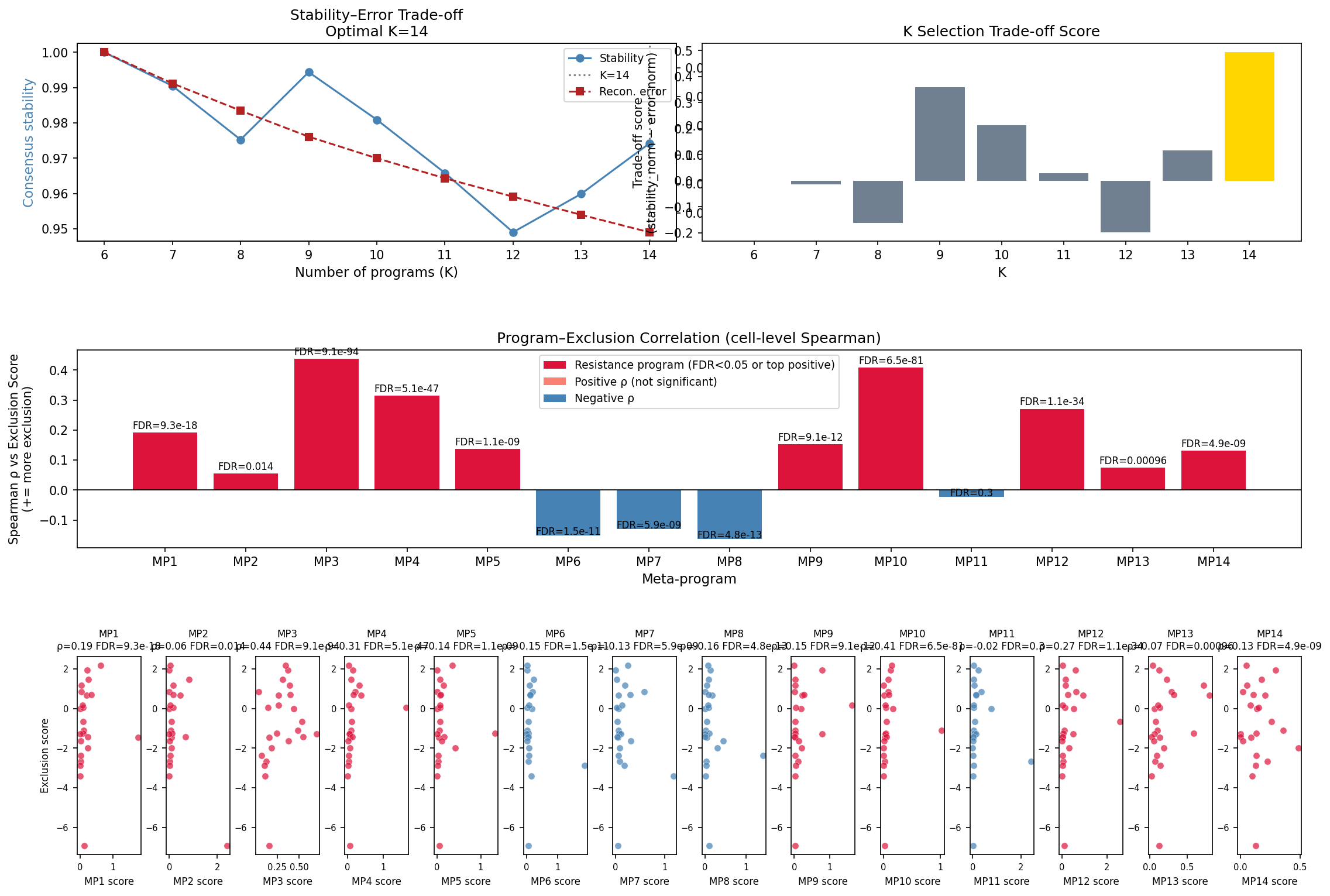
Finding 2 — Two orthogonal methods converge on the Jerby-Arnon cold-tumour axis
We attacked the malignant compartment with two computationally independent methods:
| Method | Scope | Output |
|---|---|---|
| Consensus NMF (100 seeds × K∈[6..14], stability 0.974) | Within-tumour subpopulation structure on 2,018 Mal cells | MP10: APOD, SERPINE2, SERPINA3, A2M, PMEL, MIA, CTSB, TYR, TIMP1, MT2A — patient-level Spearman ρ = 0.631 with exclusion (FDR = 0.017); cell-level ρ = 0.408 (FDR = 6.5e-81) |
| Pseudobulk DESeq2 | Between-patient counts across 23 patients | 98-gene Fisher-combined consensus (COL3A1, GLI2, WNT5A, SULF2, MECOM, GATA2, CXCL14, EDN3) emphasising ECM / Hedgehog / WNT / endothelin |
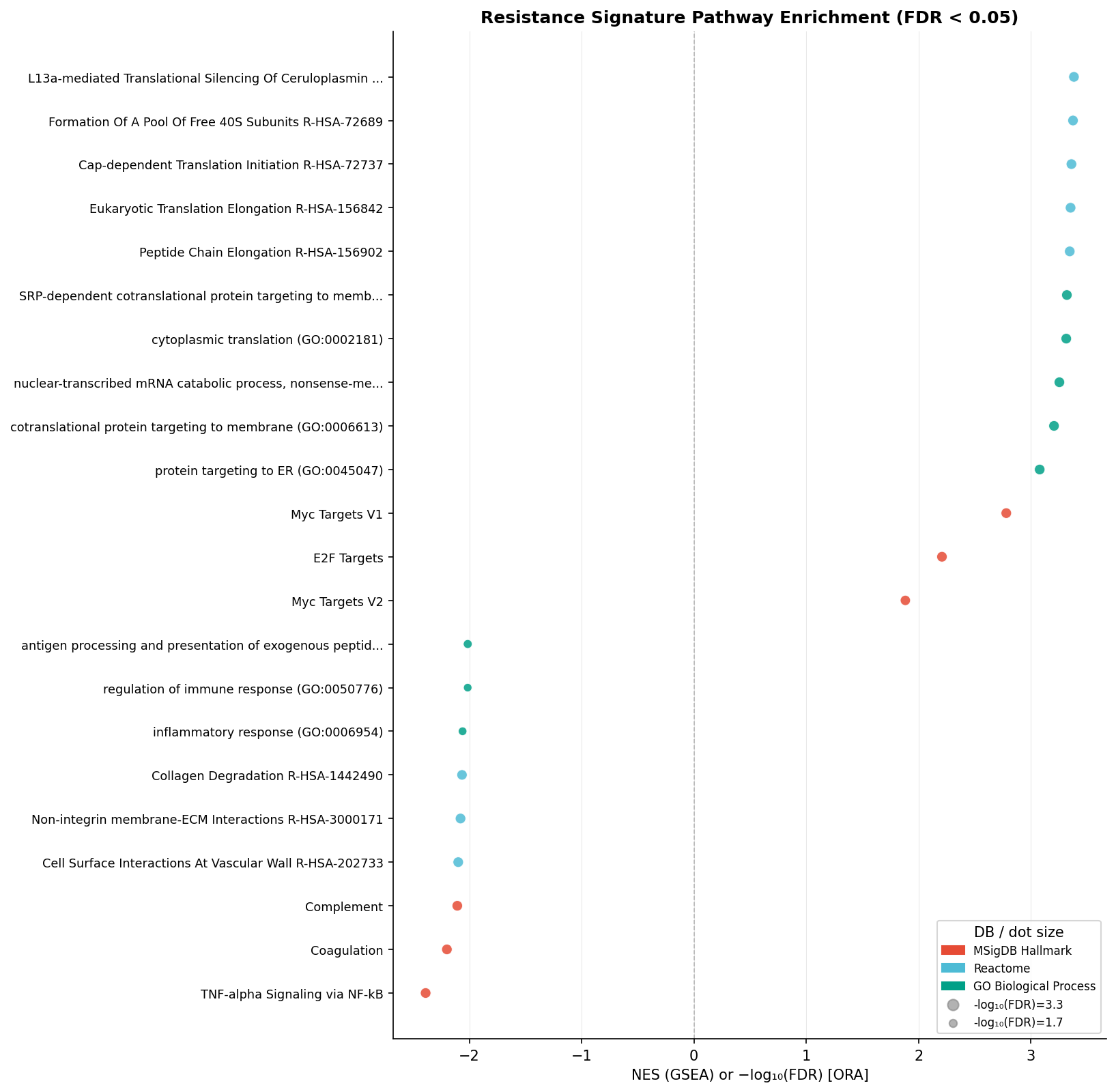
The two gene lists have zero direct overlap (hypergeometric p = 1.0). Yet projected onto the full ranked transcriptome via GSEA, both reproduce the published Jerby-Arnon exclusion programme:
"Induced module NES = +2.58, Repressed module NES = −2.51 (both p = 0.0005). 388 pathways at FDR < 0.05 with coordinated up-regulation of MYC / E2F / translation / ribosome biogenesis and down-regulation of IFN-γ response, MHC-I antigen presentation, TNF/NFκB, complement, IL-6/JAK/STAT3, and EMT."
This is a clean cross-method internal validation: orthogonal lenses agree at the pathway level even when they disagree at the gene-list level — exactly the kind of robustness signal a target-validation team needs before staking a combination programme on an axis.
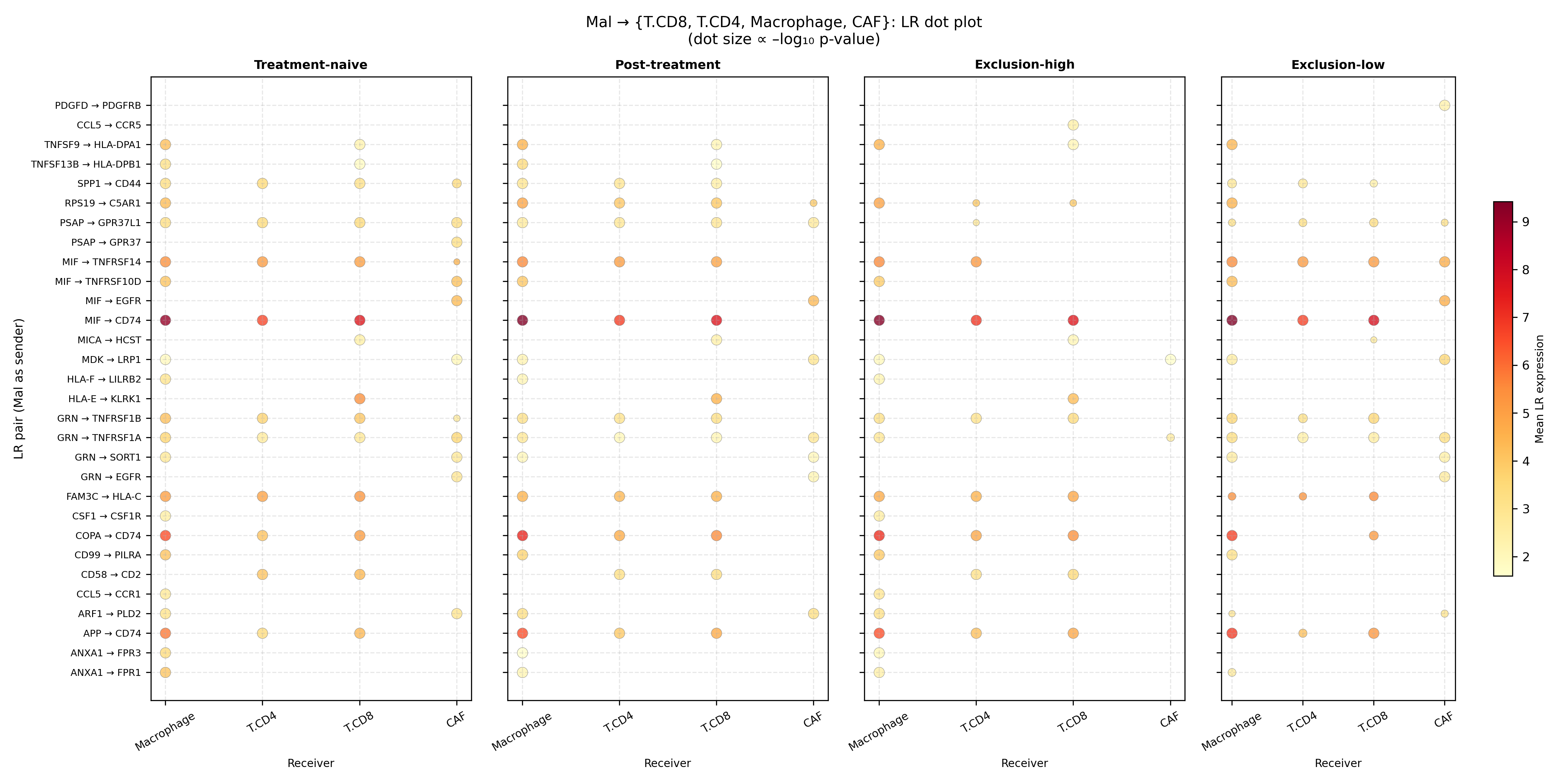
Finding 3 — A coherent malignant–immune–stromal communication network
LIANA / CellPhoneDB inference across Mal ↔ T.CD8 / T.CD4 / Macrophage / CAF (4 conditions, 1,000 permutations) returned 2,402 significant LR × cell-pair calls of 4,245 tested, with 188 constitutive across all four conditions.
Mal-as-receiver consistently outnumbered Mal-as-sender, but the dominant Mal-as-sender axis is MIF → CD74 (mean expression 7.91 across conditions), reinforced by COPA → CD74 (5.63) and APP → CD74 (5.02).
Ranking by Δ-mean enrichment in post-treatment / exclusion-high contexts surfaced 309 enriched LR pairs. The top hits form a remarkably coherent immunosuppressive network:
| Rank / theme | Pair | Sender → Receiver | Δ-excl |
|---|---|---|---|
| #1 | CD72 → SEMA4D | Macrophage → Mal | 0.93 |
| Complement evasion (4 of top 13) | RPS19 → C5aR1 | Mal → T.CD4/T.CD8/Macrophage/CAF | — |
| Galectin / metabolite | LGALS9 → SLC1A5 | — | 0.82 |
| Myeloid checkpoint | LILRB4 → LAIR1 | Macrophage → Mal | 0.85 |
| Stromal physical exclusion | COL14A1 / COL6A3 / COL12A1 → ITGA2/ITGA10 | CAF → Mal | — |
| Constitutive hub | MIF / COPA / APP → CD74 | Mal → Macrophage | dominant |
Mechanism breakdown of the 309 enriched pairs: 77 cytokine/GF, 46 ECM/CAF, 22 checkpoint, 13 chemokine, 151 other.
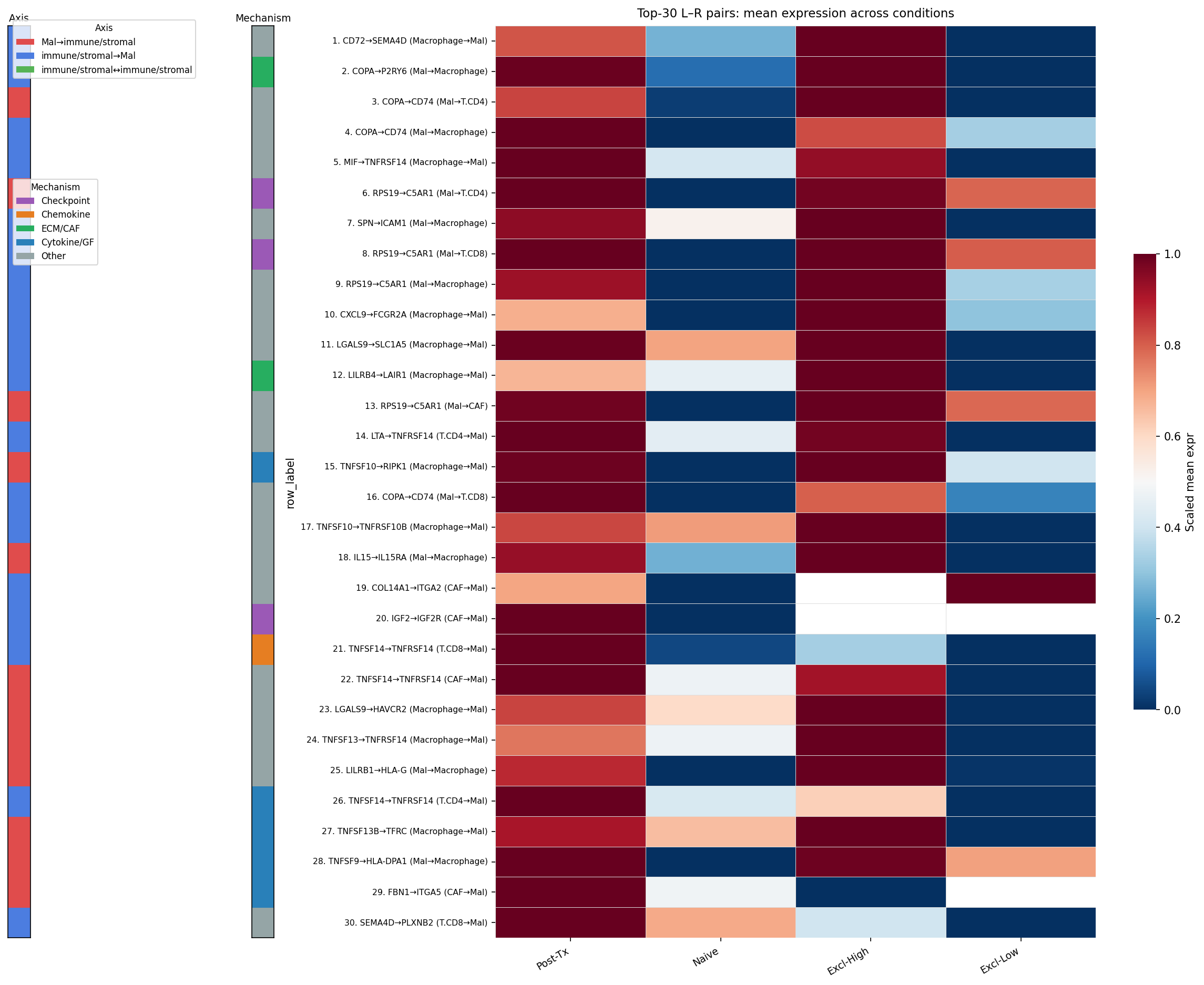
Finding 4 — Target-based repurposing yields 28 anti-PD-1 combination candidates
Querying DGIdb (≥2 sources), OpenTargets, ChEMBL and ClinicalTrials.gov against the union of MP10 + 98-gene consensus + malignant-side LR targets (39 unique druggable targets) returned 28 candidates: 24 at clinical phase ≥ 2, 9 FDA-approved, 6 with melanoma / solid-tumour data.
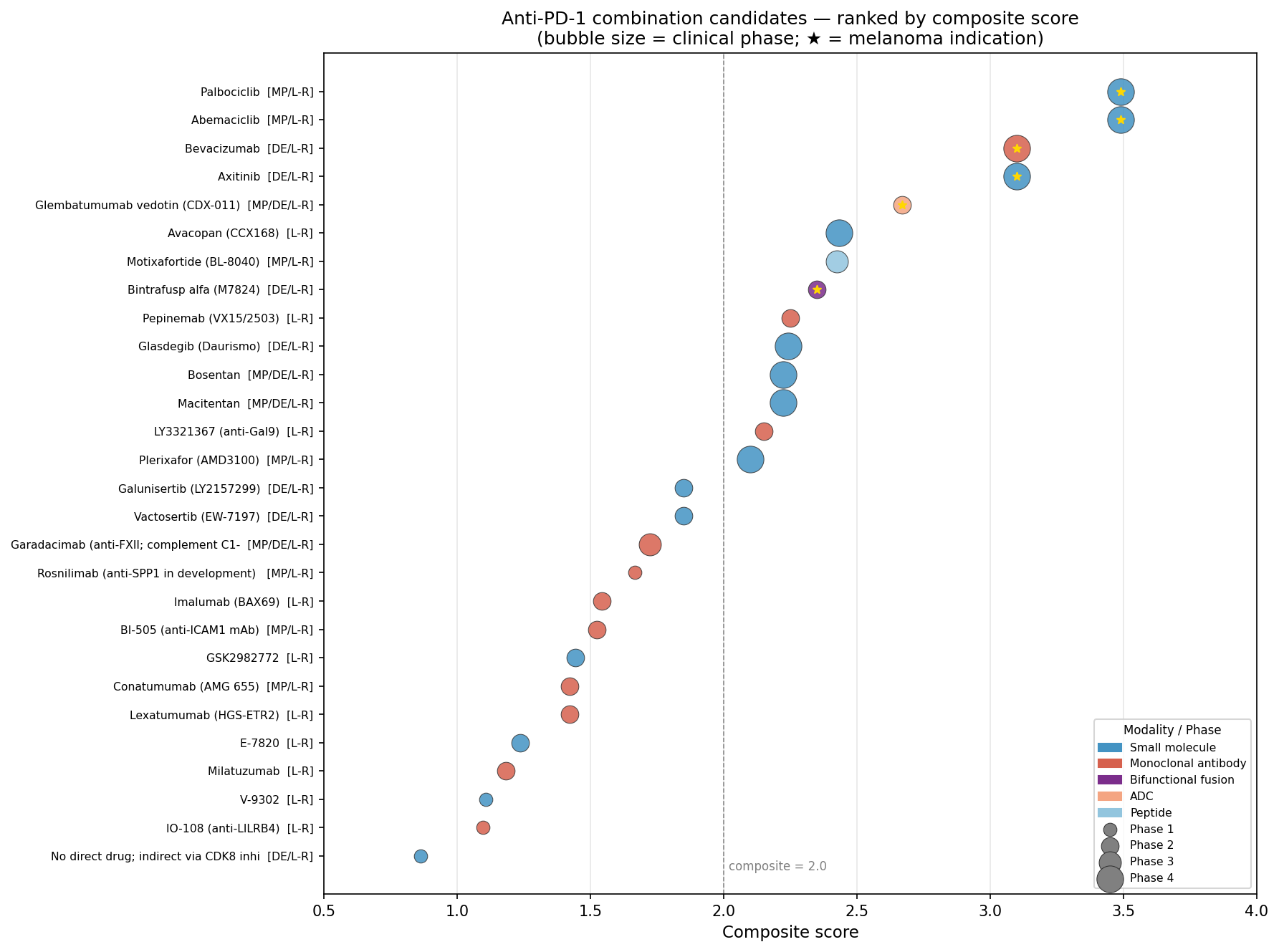
Finding 5 — Safety filtering: five tier-1 combinations, ET-axis demoted
This is where the analysis diverges most decisively from a naive "highest composite score wins" output. Cross-referencing each candidate against the curated secondary-pharmacology safety panel, OpenTargets organ-liability ledgers, IMPC knockout phenotypes, and combination-trial pharmacovigilance literature reorders the shortlist.
Tier-1 (high priority) — five anti-PD-1 combinations:
| Drug | Target | Modality | Phase | Why tier-1 |
|---|---|---|---|---|
| Bevacizumab | VEGFA | mAb | 4 | FDA-approved + anti-PD-L1 precedent (atezo/bev in HCC); manageable hypertension/GI/haemorrhage; no hepatotoxicity amplification expected |
| Glembatumumab vedotin | GPNMB | ADC (MMAE) | 2 | Direct melanoma + anti-PD-1 phase 2 (NCT02302339); GPNMB KO viable; minimal OT liabilities |
| Avacopan | C5aR1 | Small molecule | 4 | FDA-approved (ANCA vasculitis); addresses the RPS19 → C5aR1 hub (4 of top 13 LR pairs); clean safety profile |
| Pepinemab | SEMA4D | mAb | 2 | Addresses rank-1 LR pair CD72 → SEMA4D; nivolumab combination in NCT03690986 |
| Plerixafor | CXCR4 | Peptide | 4 | Extensive human safety; CXCR4 KO embryonic-lethal but adult pharmacology well-tolerated |
Demoted despite top composite scores
- Palbociclib / Abemaciclib (CDK4/6) — top composite (3.49) and the cleanest mechanistic link to the JA-induced module — demoted on hepatotoxicity grounds: ribociclib + spartalizumab Ph1 (Garrido-Castro 2025) showed grade 3/4 hypertransaminasemia in 40–47% of cohort B.
- Axitinib + pembrolizumab — KEYNOTE-426 grade 3+ ALT 13% (vs 3% sunitinib); 30.1% any-grade hepatic disorder in real-world Japanese cohort (Oya 2025). Bevacizumab preferred.
- Bosentan / macitentan — demoted despite EDN3 carrying the largest fold-change in the consensus signature because of FDA boxed warnings (EDNRA cardiac liability, hepatotoxicity).
- Bintrafusp alfa — contains an anti-PD-L1 moiety; combination with anti-PD-1 = dual checkpoint blockade with TGF-β trap; high irAE amplification risk.
Translational distance is uneven across the tier-1 list
| Candidate | Stage |
|---|---|
| Glembatumumab vedotin | Clinical evidence (direct Ph2 trial in melanoma + anti-PD-1) |
| Bevacizumab, plerixafor, avacopan | Biomarker-candidate in this disease context |
| Pepinemab | Preclinical-validation pending biomarker assays in excluded melanoma |
Why this matters for biopharma translational teams
- The exclusion-axis reframing is portable. Any single-cell IO programme that stratifies cohorts on "naive vs post-treatment" without a per-tumour infiltration metric is at risk of mixing resistant tumours with successfully infiltrated ones. The CD8/Mal log-ratio is a cheap, robust stratifier for hypothesis-generating cohorts.
- Cross-method convergence on a published axis is a stronger validation than gene-list overlap. cNMF and pseudobulk DESeq2 disagree at the gene level (overlap = 0) but converge at NES > 2.5 against the JA programme — exactly the robustness signal target-validation teams should demand before committing to a resistance axis.
- Safety-filtered repurposing flips the shortlist. The two highest composite scorers (CDK4/6i) are demoted; the largest single fold-change in the signature (EDN3) is demoted. The candidates that survive — bevacizumab, glembatumumab vedotin, avacopan, pepinemab, plerixafor — are not the ones a naive ranking would surface, and three of them map directly to specific LR pairs identified upstream.
- L-R-pair-direct combinations are auditable. Pepinemab (CD72 → SEMA4D, rank 1) and avacopan (RPS19 → C5aR1, 4 of top 13) are anchored on specific intercellular signalling events visible in the data — the kind of mechanistic provenance regulators and clinical biomarker teams reward.
Honest limitations
- Cross-sectional design — pre/post biopsies are from different patients (not matched longitudinal).
- Small N for pseudobulk DE (23 patients with malignant cells; only 2 in exclusion-low).
- Exclusion score uses raw counts, ignores NK / CD4 / Macrophage / 706 unspecified T-cells.
- CellPhoneDB is co-expression based, no multiple-testing correction across LR pairs; CAF and exclusion-low Mal cell counts are very small (45–61 and 11 cells).
- Composite score is heuristic (43 % weight on phase) — alternative weightings reorder candidates.
- Protein-level target expression has not been verified. FAERS / structural-alert pharmacovigilance was not available.
This is a hypothesis-generating scaffold, not a clinical recommendation. But it is a scaffold with internally validated biology, a defensible safety filter, and an auditable trail from a single-cell observation to a phase-2-ready combination.
Re-analysis based on GSE115978 (Jerby-Arnon L. et al., Cell 2018; PMID 30388455). Methods: Harmony integration, consensus NMF, pseudobulk DESeq2, fgsea, LIANA/CellPhoneDB, DGIdb / OpenTargets / ChEMBL / ClinicalTrials.gov target intelligence, IMPC KO + curated secondary-pharmacology safety panel.
Interested in this kind of analysis?
See how Inflexa turns single-cell atlases into safety-filtered, mechanistically anchored combination shortlists with full provenance.